- Home
- About the Journal
- Peer Review
- Editorial Board
- For Authors
- Reviewer Recognition
- Archive
- Contact
- Impressum
- EWG e.V.
Cite as: Archiv EuroMedica. 2026. 16; 1. DOI 10.35630/2026/16/Iss.1.002
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is increasingly recognized as a systemic disease with pronounced vascular and immunological involvement. Beyond acute respiratory manifestations, COVID-19 is associated with endothelial dysfunction, immune dysregulation, and coagulation abnormalities, which may persist and contribute to long-term sequelae known as Long COVID.
To systematize and analyze current evidence on immune mediated mechanisms of vascular injury in acute COVID-19 and Long COVID, with particular emphasis on endothelial dysfunction, immunothrombosis, and chronic vascular complications.
A narrative literature review was conducted based on peer reviewed clinical, experimental, and mechanistic studies published between 2019 and 2024. Data were retrieved primarily from the PubMed database and supplemented by guidelines from international organizations. The analysis focused on immune activation, cytokine storm, NETosis, endothelial injury, coagulation disturbances, and immune mediated vascular inflammation associated with SARS-CoV-2 infection.
Available evidence indicates that excessive immune activation, characterized by elevated pro-inflammatory cytokines, NETosis, oxidative stress, and direct interaction of the viral spike protein with the ACE2 receptor on endothelial cells, leads to endothelial dysfunction and disruption of hemostatic balance. These processes promote immunothrombosis, microvascular injury, and vasculitis, contributing to acute complications such as acute respiratory distress syndrome and multiorgan dysfunction. Persistent immune activation and endothelial damage appear to underlie the vascular manifestations observed in Long COVID.
SARS-CoV-2 infection induces immune mediated endothelial injury that plays a central role in both acute COVID-19 and its long-term vascular sequelae. The continuity of pathogenic mechanisms from the acute phase to Long COVID highlights the need for therapeutic strategies targeting endothelial repair, modulation of NETosis, and control of chronic immune activation.
Keywords: COVID-19, SARS-CoV-2, endothelial dysfunction, vasculitis, NETosis, cytokine storm, Long COVID
Coronavirus disease 2019 is caused by the severe acute respiratory syndrome coronavirus 2 and was declared a global pandemic by the World Health Organization on March 11, 2020 [1]. Since its emergence, SARS-CoV-2 has undergone multiple waves of global spread associated with the appearance of new variants and subvariants. Clinically, COVID-19 most often presents as an acute infectious disease with fever, dry cough, interstitial pneumonia, fatigue, headache, and loss of taste or smell [2]. In a proportion of patients, the infection progresses to severe respiratory failure with the development of acute respiratory distress syndrome. In such cases, excessive immune activation with massive release of pro inflammatory cytokines, commonly referred to as a cytokine storm, may lead to multiorgan dysfunction syndrome and increased mortality [3,4,5].
Accumulating evidence indicates that COVID-19 is not limited to viral pneumonia but represents a systemic disease with pronounced vascular involvement. Elevated levels of pro inflammatory cytokines stimulate the coagulation cascade and disrupt the physiological balance between procoagulant and anticoagulant mechanisms. In parallel, molecular mimicry and immune dysregulation contribute to the development of autoimmune and autoinflammatory processes [6]. Post infectious immune abnormalities have been documented, including persistent cytokine overproduction, hyperactivation of T lymphocytes, and increased numbers of activated macrophages, monocytes, and neutrophils [7,8,9]. These immune alterations may result in inflammatory diseases of large, medium, and small blood vessels. Autoimmune conditions reported after COVID-19 and involving the vascular system are summarized in Table 1 [10].
Table 1. Autoimmune and immune-mediated diseases associated with prior COVID-19 infection [10].
| Disease entity | Dominant disease category | Primary vascular level involved | Underlying immunological mechanism |
| Guillain–Barré syndrome | Neurological autoimmune disease | Microvascular / neural vessels | Post-infectious autoimmunity, molecular mimicry |
| Miller–Fisher syndrome | Neurological autoimmune disease | Microvascular / neural vessels | Autoantibody- mediated immune response |
| Antiphospholipid syndrome | Systemic autoimmune disease | Arterial and venous vessels | Autoantibodies against phospholipids, hypercoagulability |
| Immune thrombocytopenic purpura | Hematological autoimmune disease | Microvascular | Immune-mediated platelet destruction |
| Evans syndrome | Hematological autoimmune disease | Microvascular | Combined autoimmune cytopenias |
| Systemic lupus erythematosus (SLE) | Systemic autoimmune disease | Small and medium vessels | Immune complex deposition, complement activation |
| Kawasaki disease | Systemic vasculitis | Medium-sized arteries | Hyperinflammatory immune activation |
| Cold agglutinin disease / autoimmune hemolytic anemia | Hematological autoimmune disease | Microvascular | Autoantibody-mediated hemolysis |
| Optic neuritis | Neurological autoimmune disease | Microvascular / optic nerve vessels | T-cell–mediated demyelination |
| NMDA-receptor encephalopathy | Neurological autoimmune disease | Microvascular / CNS vessels | Autoantibodies against neuronal receptors |
| Myasthenia gravis | Neuromuscular autoimmune disease | Microvascular / neuromuscular junction | Autoantibodies against acetylcholine receptors |
| Myositis | Muscle autoimmune disease | Microvascular | Immune-mediated muscle inflammation |
| Type 1 diabetes mellitus | Endocrine autoimmune disease | Microvascular (pancreatic islets) | Autoimmune destruction of beta cells |
| Large-vessel vasculitis | Vasculitis | Large arteries | Chronic immune-mediated vascular inflammation |
| Medium-vessel vasculitis | Vasculitis | Medium-sized arteries | Immune-mediated vessel wall injury |
| Small-vessel vasculitis | Vasculitis | Small vessels | Immune complex–mediated inflammation |
| Psoriasis | Systemic inflammatory disease | Microvascular / skin vessels | Th17-driven immune dysregulation |
| Subacute thyroiditis | Endocrine inflammatory disease | Microvascular | Post-viral inflammatory immune response |
| Graves’ disease | Endocrine autoimmune disease | Microvascular | Autoantibodies against TSH receptor |
| Sarcoidosis | Systemic inflammatory disease | Microvascular | Granulomatous immune activation |
| Inflammatory arthritis | Rheumatologic autoimmune disease | Microvascular / synovial vessels | Autoimmune synovial inflammation |
Beyond the acute phase, a substantial proportion of patients develop post acute sequelae of SARS-CoV-2infection, commonly referred to as Long COVID. According to the Centers for Disease Control and Prevention, Long COVID is defined as the persistence of symptoms beyond four weeks after acute infection, whereas the World Health Organization defines it as the continuation or emergence of symptoms three months after disease onset, lasting for at least two months without an alternative diagnosis [11,12,13,14]. Long COVID is clinically heterogeneous and most frequently manifests as chronic fatigue, dyspnea, cardiac rhythm disturbances, muscle weakness, cognitive impairment, headaches, gastrointestinal symptoms, fever, and dermatological changes [15]. The risk and course of Long COVID are influenced by age, pre existing comorbidities such as asthma and obesity, overall health status before infection, and sex, with sex related differences diminishing in older age groups [16].
Importantly, growing epidemiological and clinical evidence indicates that patients with Long COVID have a higher incidence of vascular pathology, including arterial and venous thrombosis, microcirculatory dysfunction, vasculitis, atherosclerosis progression, and hypertension [11,17,18,19]. These observations suggest that persistent vascular injury and endothelial dysfunction play a central role in the long term consequences of SARS-CoV-2infection.
The relevance of this study is determined by the growing recognition of COVID-19 as a systemic vascular disease rather than an isolated respiratory infection. Despite extensive research conducted since the beginning of the pandemic, the mechanisms linking immune dysregulation, endothelial injury, and long term vascular complications remain insufficiently integrated in clinical and pathophysiological models. The increasing prevalence of Long COVID and the high incidence of vascular complications among affected patients create an urgent need for a structured analysis of immune mediated vascular injury and its clinical consequences.
The novelty of this review lies in the integrated analysis of immunological, endothelial, and coagulation related mechanisms across both the acute and post acute phases of SARS CoV 2 infection. Unlike studies focusing on isolated aspects such as cytokine storm, thrombosis, or individual autoimmune manifestations, this article synthesizes evidence on immune activation, NETosis, endothelial dysfunction, and hemostatic imbalance within a unified pathogenic framework. Particular emphasis is placed on the continuity of vascular injury from acute COVID-19 to Long COVID, highlighting persistent endothelial dysfunction as a central linking mechanism.
This article discusses well documented immunological mechanisms responsible for vascular injury during acute COVID 19 and Long COVID. Available data demonstrate that SARS-CoV-2 infection induces multidirectional activation of the immune system with profound effects on the vascular endothelium and the coagulation system. A central mechanism is the cytokine storm, which promotes endothelial dysfunction, increases vascular permeability, and activates coagulation pathways [3,4,5]. Endothelial injury is further intensified by direct interaction between the viral spike protein and the ACE2 receptor expressed on endothelial cells, leading to local inflammation, oxidative stress, and mitochondrial dysfunction [34,35].
Another critical component of COVID-19 associated vascular pathology is the formation of neutrophil extracellular traps. Enhanced NETosis has been demonstrated in both acute infection and Long COVID, where it contributes to thrombosis, sustains endothelial damage, and promotes chronic inflammation [11]. These processes are closely linked to an increased risk of cardiovascular complications, including venous and arterial thrombosis, vasculitis, and accelerated atherosclerosis [17,18,19,33]. Hemostatic abnormalities such as elevated levels of von Willebrand factor, tissue factor, plasminogen activator, and fibrin degradation products correlate with disease severity and are associated with ARDS and multiorgan failure [29,30,36–39]. Biomarkers including D dimers, cardiac troponins, and liver enzymes have demonstrated prognostic value for severe disease and mortality [43–46].
The objective of this study is to analyze and systematize current evidence on immune mediated mechanisms of vascular injury in COVID-19 and Long COVID. The specific tasks of the review include the following. To describe the physiological role of the vascular endothelium in immune regulation and coagulation. To characterize the key immunological processes activated during SARS-CoV-2 infection that contribute to endothelial damage. To evaluate the role of cytokine storm and NETosis in the development of thrombosis and vasculitis. To summarize evidence on coagulation disturbances and their clinical significance. To assess the contribution of persistent immune activation to vascular complications observed in Long COVID.
Taken together, persistent endothelial dysfunction and chronic immune activation appear to represent key mechanisms underlying both acute vascular complications of COVID-19 and the long term manifestations observed in Long COVID. Although antithrombotic prophylaxis during the acute phase is addressed in current ISTH guidelines, evidence based strategies targeting chronic vascular and immunological disturbances in Long COVID remain insufficient. Further research is required to develop therapeutic approaches focused on endothelial repair, modulation of NETosis, and attenuation of sustained inflammatory responses.
Table 2. Immunological mechanisms of endothelial injury and vascular complications in COVID 19 and Long COVID
| Patho- physiological mechanism | Key mediators and processes | Type of endothelial or vascular injury | Associated clinical manifestations | Evidence source references |
| Cytokine storm | IL 6, IL 1β, TNF α, macrophage and monocyte hyperactivation | Endothelial dysfunction, increased vascular permeability | ARDS, MODS, acute respiratory failure, acute thrombosis | [4,5,22,23] |
| NETosis | Neutrophil extracellular traps, activated neutrophils, impaired NET clearance | Microvascular injury, immuno- thrombosis | Microthrombosis, organ perfusion disorders, vascular manifestations of Long COVID | [7,11,19] |
| Direct viral endothelial injury | SARS CoV 2 spike protein, ACE2 receptor binding, oxidative stress | Endothelialitis, mitochondrial dysfunction | Vasculitis, microcirculatory failure, organ ischemia | [34,35,36] |
| Coagulation cascade dysregulation | von Willebrand factor, tissue factor, PAI 1, elevated D dimer | Prothrombotic state, thrombotic microangiopathy | Venous and arterial thrombosis, pulmonary embolism, aortic thrombosis | [37,38,39] |
| Persistent immune activation in Long COVID | Chronic inflammation, autoantibodies, sustained NETosis | Persistent endothelial dysfunction | Chronic fatigue, hypertension, cognitive impairment, vascular Long COVID | [12,17,40] |
This article is based on a narrative literature review focused on immunological mechanisms of vascular injury associated with acute COVID 19 and Long COVID. The analysis included clinical studies, experimental and mechanistic research, narrative and systematic reviews, and meta analyses addressing endothelial dysfunction, immune activation, NETosis, coagulation abnormalities, and immune mediated vascular inflammation in the context of SARS CoV 2 infection.
Literature searches were performed primarily using the PubMed database. Additional information was obtained from official documents and guidelines published by the World Health Organization, the Centers for Disease Control and Prevention, and the International Society on Thrombosis and Haemostasis. The search strategy included combinations of the following keywords: COVID 19, SARS CoV 2, endothelial dysfunction, vasculitis, immunothrombosis, NETosis, coagulation, Long COVID, vaccine, and vaccination.
Publications from 2019 to 2024 were considered, without geographical restrictions. Both human studies and experimental mechanistic research were eligible for inclusion.
Inclusion criteria were as follows. Peer reviewed publications presenting clinical, experimental, or mechanistic data on immune mediated endothelial injury, vascular inflammation, thrombosis, coagulation disturbances, or post acute vascular complications associated with SARS CoV 2 infection. Studies addressing Long COVID with documented vascular, endothelial, or immunothrombotic involvement. Reports describing vasculitis or immune mediated vascular reactions temporally associated with COVID 19 vaccination, included for comparative and contextual analysis.
Exclusion criteria included publications lacking relevance to vascular or endothelial pathology, studies focused exclusively on non vascular organ systems without immunological or endothelial implications, isolated case reports without mechanistic discussion, non peer reviewed opinion pieces, and articles with insufficient methodological description.
Reference lists of selected publications were screened manually to identify additional relevant sources. Given the narrative nature of the review, no formal systematic review protocol, quantitative synthesis, or risk of bias assessment was applied.
Ethical approval was not required, as the study did not involve original patient data or experimental interventions.
The primary functions of vascular endothelial cells include: barrier function (highly selective and regulating inflammatory and immune responses), transport function (responsible for intercellular communication and pinocytosis), reparative function (both structural and functional), angiogenesis (adaptive depending on the type of injury), regulation of coagulation (supporting natural blood flow and preventing unintended clot formation), adjustment of vessel lumen diameter (vasodilation or vasoconstriction depending on stimuli), metabolic function (responsible for the synthesis of growth factors, adhesion molecules, and receptors), and immunological function (responding to a wide spectrum of immune cells, exhibiting histological compatibility with antigens, and regulating antigen-presenting cells) [20]. The endothelium forms the inner lining of arteries, veins, and capillaries, thereby maintaining direct contact with circulating blood, its components, and surrounding tissues [21]. In addition to its fundamental role in enabling substrate distribution to tissues, the endothelium also performs endocrine and paracrine functions, demonstrating high tissue specificity and the ability to adapt to changing physiological conditions [22]. Regulation of inflammation is achieved through a variety of external signals and intracellular mediators. External mediators include anti-inflammatory cytokines, transforming growth factor β (TGF-β), interleukin-10 (IL-10), the interleukin-1 receptor antagonist (IL-1Ra), and high-density lipoprotein (HDL). A key factor contributing to inflammatory homeostasis is the production of nitric oxide in response to stressors arising from endothelial injury [23,24]. Control of coagulation processes is possible due to natural anticoagulants, platelet inhibitors, and fibrinolytic proteins, which together ensure proper organ perfusion. Since tissue factor released from injured vessel walls or circulating monocytes plays a central role in the development of thrombotic atherosclerotic lesions in coronary arteries, particular attention has been given to its surface antagonist—tissue factor pathway inhibitor (TFPI) [25]. Other important surface proteins include protein C, protein S, protein Z, nitric oxide, glycosaminoglycans, β2-glycoprotein I, tissue plasminogen activator, thrombomodulin, and urokinase-type plasminogen activator [26,27,28].
In cases of acute respiratory distress syndrome (ARDS), COVID-19 infection causes both endothelial injury and an inflammatory response characterized by activation of immune cells, leading to the release of large quantities of pro-inflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor α (TNF-α). In severe forms of the disease, excessive activation of the immune system results in massive cytokine release, known as a “cytokine storm”. The cytokine storm activates coagulation pathways, and the exaggerated response to ongoing infection disrupts the balance between pro- and anticoagulant mechanisms, resulting in microthrombus formation, disseminated intravascular coagulation (DIC), and ultimately multiorgan failure [29,30] (Figure 1). The exceptionally intense and persistent systemic inflammatory state contributes to numerous complications, including cardiovascular, metabolic, neurodegenerative, musculoskeletal, oncological disorders, and depression. These conditions may also underlie symptoms observed in Long COVID (LC) and contribute to ongoing organ damage [30,31,32]. Neutrophils also play an important role in the acute inflammatory response by forming neutrophil extracellular traps (NETs). Studies have indicated that persistent NET release or impaired NET clearance following COVID-19 resolution may contribute to the pathogenesis of LC. Additionally, NETosis has been shown to remain elevated in patients with LC compared with control groups [11].
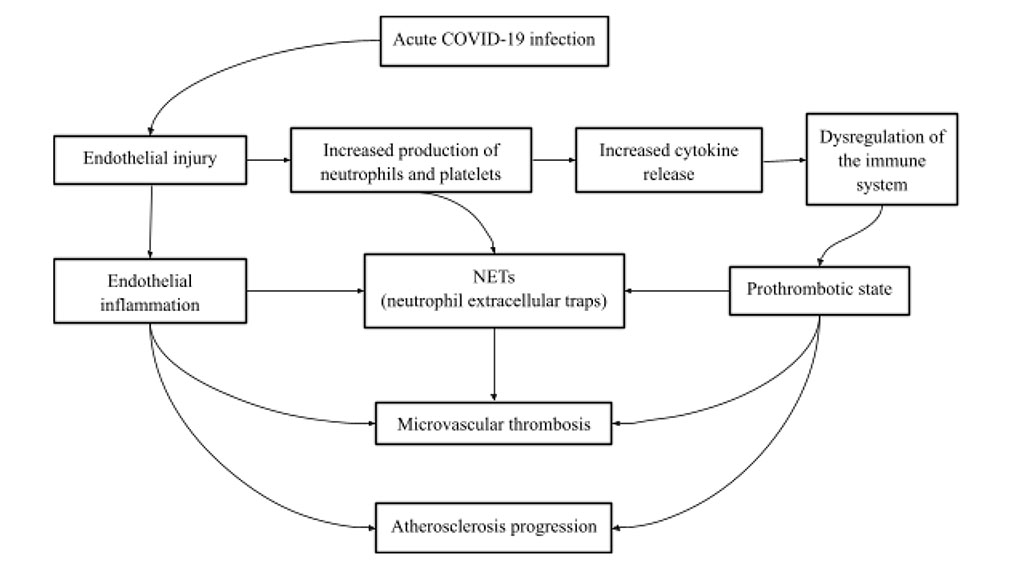
Figure 1. Diagram illustrating the processes from COVID-19 infection to microvascular thrombosis and accelerated atherosclerosis
Cardiovascular complications are common among patients with Long COVID (LC), largely due to the predominance of prothrombotic and pro-adhesive processes within vessels altered by the disease. Inflammatory vascular syndromes have been demonstrated in patients with COVID-19, confirmed histologically in the liver, lungs, skin, and kidneys [32]. The main pathophysiological changes were observed in small and medium-sized arteries and were described as acute endothelialitis, which may progress into hypersensitive acute vasculitis [33]. Several mechanisms have been proposed to directly contribute to vascular endothelial cell injury. The first involves the binding of angiotensin-converting enzyme 2 (ACE2), expressed by endothelial cells, to the SARS-CoV-2 spike protein during infection, which triggers inflammatory activation and consequently leads to progressive structural and functional damage to small and medium-sized vessels [34]. A second potential mechanism is the collateral effect of type I interferons released in response to infection, which may induce mitochondrial dysfunction [35]. NET-associated mediators also contribute to enhanced pro-inflammatory activity, promoting the recruitment of immune cells into atherosclerotic plaques, ultimately driving the progression of atherosclerosis [11].
COVID-19 infection alters multiple processes within the coagulation cascade—levels of complement, plasminogen activator inhibitor-1 (PAI-1), and von Willebrand factor (vWF) are elevated, leading to increased expression of tissue factors in response to inflammatory cytokines, microvascular dysfunction, and the promotion of thrombotic phenomena that occlude the lumina of local vessels [36,37,38,39]. (Figure 2)
![Figure 2. Possible sequence of changes leading to vascular thrombosis and worsening of aortic aneurysms [40].](artikel-2-bild-2.jpg)
Figure 2. Possible sequence of changes leading to vascular thrombosis and worsening of aortic aneurysms [40].
Based on the understanding of the pathophysiological mechanisms accompanying COVID-19 infection, it can be concluded that the predominance of prothrombotic processes, endothelial dysfunction, and alterations in blood parameters (increased concentrations of pro-inflammatory cytokines and hyperactivation of immune cells) ultimately lead to vascular thrombosis, while also increasing the risk of thrombus formation in large vessels such as the aorta [40].
In the latest ISTH guidelines on antithrombotic prophylaxis, separate recommendations have been developed for specific patient groups: non-hospitalized, non–critically ill hospitalized, critically ill hospitalized, and post-discharge patients. For non-hospitalized patients, sulodexide is recommended, as it has been shown to reduce the risk of hospitalization and the potential need for oxygen therapy. In non–critically ill hospitalized patients, a prophylactic dose of low molecular weight heparin or unfractionated heparin (LMWH/UFH) is advised, as it has been demonstrated to reduce mortality compared with patients who did not receive antithrombotic prophylaxis. Among critically ill hospitalized patients, a prophylactic dose of LMWH/UFH is recommended to prevent venous thromboembolism (VTE) in individuals without active bleeding or high bleeding risk. Patients discharged from the hospital may still have an elevated risk of thrombotic events; therefore, prophylactic doses of rivaroxaban (a DOAC) are recommended [41]. Additionally, the literature mentions supportive effects of preparations containing rutosides in the management of venous insufficiency, as they strengthen vascular integrity and provide anti-edematous effects. Oxerutins have also demonstrated anti-exudative properties by reducing capillary permeability. Pharmacotherapy should continue for at least 50 days. In cases of pronounced microcirculatory overload, treatment may be supplemented with compression therapy (following Doppler ultrasound assessment) and lymphatic drainage [42]. In patients with COVID-19, biochemical, inflammatory, and coagulation biomarkers are useful in assessing disease severity and prognosis. Among the most valuable biochemical markers are D-dimers and cardiac markers (troponins). Markers of muscle damage, particularly myocardial injury, were significantly elevated in patients with severe and fatal disease courses. In cases progressing to multiorgan failure (MOF), liver enzymes—alanine aminotransferase (ALT) and aspartate aminotransferase (AST)—were markedly increased, and critical abnormalities were found in renal function parameters (creatinine, blood urea nitrogen) [43,44].
Liver function appears to be an important predictor of mortality in patients with COVID-19. Notably, AST levels have been found to correlate strongly with mortality risk compared with other parameters [45].
Elevated inflammatory biomarkers indicate widespread vascular inflammation and disturbances in the coagulation system, which may impair the function of vital organs. C-reactive protein (CRP) levels were increased in patients in the early stages of severe disease, even preceding detectable abnormalities on computed tomography. CRP has been linked to disease progression and represents an early predictor of severe clinical course [46].
COVID-19 vaccinations, particularly those based on mRNA technology (e.g., Pfizer-BioNTech, Moderna), have revolutionized the global response to the pandemic, yet they have also been associated with certain adverse immune reactions, including cases of vaccine-induced vasculitis. Studies indicate that these events may result from an autoimmune response triggered within the host organism [47]. Although post-vaccination vasculitis is considered a rare occurrence, reports suggest that it may affect small and medium-sized vessels or manifest as cutaneous vasculitis. Additionally, systemic symptoms such as joint pain and fever have been observed [47]. A review analyzing cases of vasculitis following COVID-19 vaccination identified nearly 158 documented instances, with the most frequently reported forms being IgA-associated vasculitis and ANCA-positive vasculitis [47]. Management of vaccine-induced vasculitis requires prompt diagnosis and initiation of immunosuppressive therapy, including corticosteroids and antiviral agents in cases where viral activity is implicated. It is crucial to closely monitor patients for potential relapses of vasculitis, particularly in individuals with a predisposition to autoimmune reactions, as inadequate control may lead to long-term health complications [48].
The findings of this review underscore the central role of the vascular endothelium in the pathogenesis of both acute SARS-CoV-2 infection and its long term sequelae, including Long COVID. Accumulated evidence indicates that an excessive inflammatory response, characterized by elevated levels of IL-6, IL-1β, and TNF-α, leads to endothelial dysfunction and initiates a cascade of disturbances in the coagulation system, ultimately impairing organ perfusion [3,4,5,6,29]. A key mechanism in this process is the cytokine storm, which not only activates coagulation pathways but also directly damages endothelial cells and disrupts the physiological balance between procoagulant and anticoagulant factors [29,30]. Clinical severity of COVID-19 has been shown to correlate with biomarkers of endothelial injury, including D-dimer, cardiac troponins, and liver transaminases [43–46], which is consistent with clinical observations linking endothelial damage to the risk of ARDS and MODS [3–5].
The analysis further highlights the growing significance of NETosis in the development of both acute and chronic vascular complications. Increased formation and persistence of neutrophil extracellular traps, observed in acute COVID-19 as well as in Long COVID, contribute to sustained inflammation, progression of atherosclerotic lesions, and an increased risk of venous and arterial thrombosis [11,17–19,33]. These observations support the concept that SARS-CoV-2 infection may not only activate immune responses typical of acute viral infections but can also initiate a chronic autoinflammatory process, resulting in persistent endothelial dysfunction even after resolution of the acute clinical phase.
Another important component of COVID-19 associated vascular pathology is the direct interaction between the SARS-CoV-2 spike protein and the ACE2 receptor expressed on endothelial cells. This mechanism, well documented in experimental and clinical studies [34], facilitates viral entry and simultaneously triggers localized inflammation, oxidative stress, and mitochondrial dysfunction within the endothelium [35]. These data support the view that endothelial injury in COVID-19 arises from a combination of direct viral effects and secondary immune mediated mechanisms.
With respect to coagulation abnormalities, multiple studies have demonstrated elevated levels of prothrombotic mediators, including von Willebrand factor, tissue factor, and plasminogen activator [36–39]. Such alterations promote the development of thrombotic microangiopathy, which plays a critical role in organ damage and vascular complications observed in both acute COVID-19 and Long COVID [37–40]. In addition, histologically confirmed cases of vasculitis involving the lungs, skin, liver, and kidneys in patients with COVID-19 further support the immunological basis of vascular injury in this disease [32,33].
The clinical relevance of these findings is reflected in current therapeutic strategies. According to ISTH guidelines [41], appropriate antithrombotic prophylaxis reduces the incidence of thrombotic complications and mortality, particularly in hospitalized patients. However, evidence based recommendations for the management of persistent vascular dysfunction in Long COVID remain limited. This highlights the need for further studies aimed at developing therapeutic approaches focused on endothelial repair, modulation of NETosis, and attenuation of chronic inflammatory activity.
An additional but less frequent aspect of immune mediated vascular pathology includes reported cases of vasculitis following COVID-19 vaccination, most commonly IgA mediated and ANCA positive forms [47,48]. Although such events are rare, they suggest that immune activation may unmask or trigger autoimmune vascular reactions in predisposed individuals. These observations do not challenge the overall benefit of vaccination but emphasize the importance of continued pharmacovigilance and identification of populations at increased risk.
Taken together, available evidence characterizes COVID-19 as a multisystem disorder in which endothelial dysfunction, persistent immune activation, and hemostatic imbalance are central pathogenic features. These mechanisms not only influence the severity and outcome of the acute infection but also contribute to the development of long term vascular complications that define the clinical spectrum of Long COVID [3,4,5,6,29].
This review has several limitations that should be acknowledged. First, due to its narrative design, the selection and interpretation of literature were not based on a formal systematic review protocol, and no quantitative synthesis or risk of bias assessment was performed. Second, the included studies were heterogeneous with respect to study design, patient populations, disease severity, and outcome measures, which limits the direct comparability of findings. Third, much of the available evidence on vascular and immunological mechanisms in Long COVID is derived from observational and cross sectional studies, while longitudinal data remain limited. Finally, reported associations between immune activation, endothelial dysfunction, and clinical outcomes do not in all cases establish direct causality, underscoring the need for prospective mechanistic and interventional studies [41,43].
SARS-CoV-2 infection is associated with multilevel activation of the immune system and pronounced endothelial dysfunction, which together constitute the pathophysiological basis of both the acute phase of COVID-19 and its long-term sequelae observed in Long COVID. Cytokine storm, oxidative stress, enhanced NETosis, and direct interaction of the virus with the ACE2 receptor expressed on endothelial cells represent key mechanisms of vascular injury. These processes initiate a cascade of coagulation disturbances, leading to increased procoagulant activity, thrombotic microangiopathy, and a higher risk of organ related vascular complications. Persistent immune activation in combination with sustained endothelial dysfunction appears to play a central role in the development of the long-term vascular manifestations of Long COVID.
Antithrombotic prophylaxis according to current ISTH recommendations remains an essential component of management in hospitalized patients with acute COVID-19. However, evidence based therapeutic strategies targeting chronic vascular and endothelial dysfunction in Long COVID are still lacking. Although vasculitis following COVID-19 vaccination has been reported only rarely, these cases indicate that immune mediated vascular reactions may occur in predisposed individuals, underscoring the importance of continued safety surveillance.
In summary, COVID-19 should be regarded as a systemic disease with significant and persistent vascular consequences extending beyond the acute phase of infection. Further research focused on endothelial repair, modulation of NETosis, and control of chronic immune activation is required to improve long-term outcomes and reduce the vascular burden associated with SARS-CoV-2 infection.
All authors contributed substantially to the conception and design of the study, literature analysis and interpretation, drafting and critical revision of the manuscript, and approved the final version for publication.
Conceptualization and methodology: Urszula Chmielecka, Olga Wcisłek
Literature review and data extraction: Urszula Chmielecka, Olga Wcisłek, Julia Wendt, Dominika Raether, Aleksandra Markuszewska, Agnieszka Anna Bugała, Adam Andrzejewski
Writing - original draft preparation: Urszula Chmielecka, Olga Wcisłek, Julia Wendt, Dominika Raether, Aleksandra Markuszewska, Agnieszka Anna Bugała, Adam Andrzejewski
Writing - review and editing: Olga Wcisłek, Urszula Chmielecka, Julia Wendt, Dominika Raether, Aleksandra Markuszewska, Agnieszka Anna Bugała, Adam Andrzejewski
The authors declare that no artificial intelligence tools were used in the generation, writing, editing or revision of this manuscript. All content was created solely by the authors.
The article did not receive any funding.
Authors declare no conflicts of interest.

|
||